Neural networks for simulating molecular movement * The field of machine learning makes it possible to apply quantum mechanics effectively in molecular simulations
[Translation by Dr. Nachmani Moshe]
A new study led by researchers from the University of North Carolina and the University of Florida shows that it is possible to "teach" artificial neural networks to apply the laws of quantum mechanics in order to describe the movements of molecules, that is, to develop simulations used in a wide range of fields of knowledge.
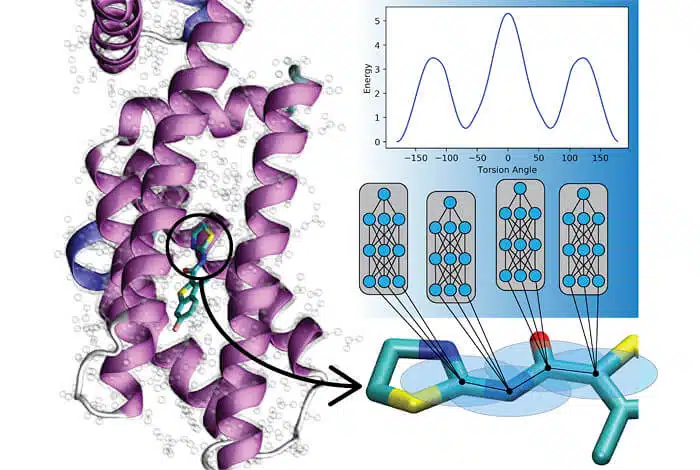
"The significance of the research findings is that we are now able to model materials and molecular dynamics at a billion times faster rate compared to normal quantum methods, while maintaining the same level of accuracy," said one of the researchers. Understanding how molecules move is essential and valuable for drug development, protein simulations and chemistry of reactive substances, for example, and both methods of quantum mechanics and experimental (empirical) methods can be utilized within these simulations.
The new method holds the promise of advancing the capabilities of researchers in many fields and improving machine learning-based accuracy in future studies of metal alloys and explosives physics. Algorithms of quantum mechanics, used in ordinary computers, can describe with great precision the mechanical movements of a compound in a defined environment. However, quantum mechanics methods are limited in terms of varying molecular size of the compounds, and their simulations are therefore less accurate. Even a small increase in molecular size within a given simulation can drastically increase the computing power required. Therefore, scientists often use empirical information, which describes the movement of atoms in the framework of classical physics and Newton's laws, which give rise to simulations that are also suitable for billions of atoms or millions of chemical compounds. Traditionally, empirical potentials are required to weigh tradeoffs between obsolescence (of a model) and accuracy - when many parameters are properly tuned for one compound, the accuracy for other compounds is low. In contrast, the researchers developed a machine learning approach called "transfer learning" that allows researchers to develop empirical potentials by learning from data collected about millions of compounds. The new approach can be applied to new molecules at the level of milliseconds, a result that will allow the study of many more compounds in much shorter times.
The article describing the study

4 תגובות
Science is not usually done by one person. If there is a breakthrough discovery others begin to examine its various implications immediately.
Professor Shashua theoretically proved that a neural network functions according to quantum mechanics over time. This is no exaggeration.
The result is not new. Professor Shashua Amnon published a third groundbreaking article on the way this is done.
It was a year ago.
The original theoretical discovery is perhaps his.
No matter how true and how related it is, adding "quantum physics" to anything makes it sound excessive.