A new discovery of genetic mutations provides clues about how the disease brutally destroys motor nerve cells and robs people of their ability to move. These findings may lead to new drugs for a disease that has long eluded treatment
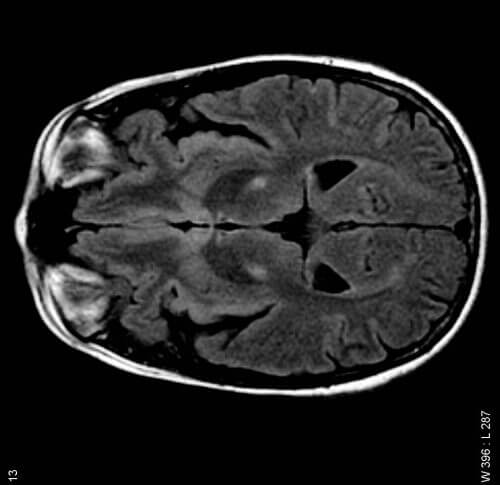
- Amyotrophic lateral sclerosis (ALS), a degenerative disease of the nervous system known as Lou Gehrig's disease, attacks nerve cells that extend from the brain and spinal cord to muscles throughout the body.
- Sophisticated methods for determining DNA sequences have led to many discoveries about the genetic basis of ALS. Studies show that changes in many genes increase the risk of getting sick.
- Gene silencing using a synthetic molecule called an antisense oligonucleotide is a possible treatment for certain types of ALS. Researchers will also look for ways to measure disease progression to help with early diagnosis and drug development.
amyotrophic lateral sclerosis (ALS) strikes without warning. The disease, which robs nerve cells of their ability to communicate with the muscles of the body, begins painlessly, with mild initial symptoms, such as stumbling, clumsy behavior or slurring words, which are easy to ignore. The disease itself did not receive much public interest until the legendary baseball player Lou Gehrig of the New York Yankees, began dropping balls and collapsing on the field for no apparent reason. Gehrig, nicknamed the Iron Horse because he played 2,130 consecutive games over 14 years, was diagnosed with ALS in June 1939. He gave a touching farewell speech at Yankee Stadium the following month. Gehrig's mobility deteriorated so rapidly that by December he was already too weak to participate in the ceremony where he was inducted into the American Baseball Hall of Fame. The increasing paralysis eventually confined him to his bed. He died in June 1941 when he was 37 years old.
Today, more than 6,000 patients are diagnosed in the US each year with ALS, known as Lou Gehrig's disease in the US and motor neuron cell disease in Europe. The disease most often affects people aged 50 to 60, but it can start much earlier and even later, over the age of 80. With the onset of the disease, nerve cells in the brain and spinal cord begin to die. Since these cells send signals from the brain through the spinal cord to the muscles, their death impairs the ability to move, fine motor skills, speech and even swallowing. In most cases, the higher faculties of the brain are not affected: people with ALS are forced to watch their bodies collapse as the disease progresses relentlessly. Soon they are confined to a wheelchair, and eventually to their bed. Unable to communicate, eat or breathe on their own, most die of respiratory failure within three to five years. The only drug approved for the treatment of ALS by the US Food and Drug Administration (FDA) is riluzole containing an inhibitory substance Glutamate, and extends survival by three months on average. There is no cure for the disease.
The pioneering neurologist Jean-Martin whistled, who identified the disease in 1869, chose a name that describes the symptoms of the disease: "Sclerosis" is hardening or scarring that occurs due to the process of nerve degeneration; "Amyotrophic" means a lack of nourishment to the muscles and "lateral" due to the area in the spinal cord where some of the motor neurons that die are found. Despite Charco's clear characterization, nearly 150 years later the complexity of ALS continues to challenge researchers. Although the disease is always fatal, about 10% of patients survive more than 10 years, and some even longer, and no one knows why. This minority includes physicist Stephen Hawking, who has lived with ALS for more than 50 years. Recent research suggests that environmental factors play only a small role in the onset of ALS, perhaps by increasing the vulnerability of people who are already genetically predisposed to the disease. The biggest puzzlement is that the disease attacks mostly at random. Less than 10% of cases are due to genetic traits that pass from generation to generation in the family. The rest of the cases are defined as non-hereditary, or random.
During the last decade, sophisticated methods for determining DNA sequences have led to a rapid increase in the understanding of the biology underlying the disease. Studies suggest that many genes, acting individually or together, can increase the tendency to get sick. There is a connection between specific mutations and almost 70% of familial cases, and about 10% of random cases. The abundance of new genetic data paves new and promising paths to better treatments. Gene silencing is one possible treatment for some types of ALS. Two drugs targeting two separate genes are slated for clinical trials in 2017. At the same time, researchers are identifying biological markers, including substances that can be measured in body fluids or electrical activity in the brain, which could help doctors diagnose the disease earlier and better assess its progress. Such markers can also be useful in the development of other drugs.
Early genetic clues
Although people with familial ALS, most of whom have a 50% chance of passing the disease on to the next generation, represent a small portion of ALS sufferers, they have played a much larger role in deciphering the genetic basis of the disease. The first genetic link to ALS was obtained in 1993 from studies that identified a mutation in the gene called SOD1 in approximately 20% of familial ALS cases. The SOD1 gene encodes an antioxidant enzyme, superoxide dismutase, which makes the molecule very active Superoxide, a free radical of oxygen, into less harmful forms.
Researchers first hypothesized that the mutation in SOD1 weakens the enzyme's antioxidant activity and therefore allows oxygen free radicals to wreak havoc on motor neurons. A quarter of a century later we have learned with almost absolute certainty that this is not the case. On the contrary, the mutation seems to cause excess toxic activity, where the enzyme does something beyond its normal functions.
The new role leads to changes in the spatial shape of certain proteins in nerve cells. Most autopsies of people with ALS show typical brain pathology: lumps of protein that accumulate inside motor neurons. In order for these nerve cells to function optimally, it is necessary to efficiently recycle the building blocks of the proteins, but in ALS the circulation system is impaired. All proteins, including enzymes, need to fold into precise three-dimensional shapes during their formation in cells so that they can function as required. Researchers eventually discovered that mutations likely cause proteins to misfold and form clumps. As part of cellular activity, cells mark the damaged proteins with a so-called molecular marker Ubiquitin, which signals that it is necessary to eliminate them. When the cellular circulatory system is overwhelmed and the waste accumulates. In people with certain types of familial ALS, motor neurons become filled with clumps of defective SOD1 proteins that are marked with ubiquitin.
A major breakthrough in ALS research occurred in 2006, when scientists looked at ALS cases that were not characterized by mutations in 1SOD. They discovered a different protein in almost all of them, TDP-43, which also formed clumps in motor neurons. TDP-43 belongs to a group of proteins that control the activity of molecules Messenger RNA: Mobile copies of the DNA that serve as templates for the production of the proteins encoded by the DNA "letters". TDP-43 binds to the messenger RNA, directs its processing in the nucleus, leads it to its destination in the cell and performs other important roles in the "translation" of the information stored in the RNA into protein output. For some reason in ALS, TDP-43 proteins are pulled out of the nucleus and begin to accumulate in the surrounding cytoplasm. It is even possible that the proteins act as a kind of magnet that attracts more copies of the protein to the cytoplasm. Scientists have not yet discovered whether the mutations in the TDP-43 gene cause the loss of activity of the protein (because it is removed from the nucleus) or a toxic excess activity (because it accumulates in the cytoplasm), or both.
The identification of TDP-43 as the main protein that accumulates in most ALS cases helped geneticists zero in on the coding gene, TARDBP, where they discovered rare mutations in some families with the hereditary form of the disease. The most influential discovery in the research is the understanding that changes in the RNA-binding protein can cause ALS. After that, researchers identified several additional genes that cause ALS and encode proteins involved in RNA control, and they believe there will be many more. In the late 2000s there was an explosion of genetic discoveries in the field of ALS, and every year one or two genes related to the disease emerged. But the most exciting discovery has yet to be made.
DNA repeats are out of control
The findings emerged from studies of several families with a hereditary form of ALS. In 2011, two groups of scientists independently reported the discovery of a strange mutation in a gene with a strange name as well - C9ORF72, which means the 72nd open reading frame (the coding part of the gene), found on chromosome 9. In healthy people, this gene includes a short sequence of DNA - GGGGCC - that is repeated two to 23 times. In people with the C9ORF72 mutation, this segment is repeated hundreds or even thousands of times.
Additional studies have suggested that these excess recurrences can explain 4% to 50% of familial ALS cases and 5% to 10% of apparently random cases. Interestingly, the discovery of the mutations provides a genetic link between ALS and another disease, a type of dementia called frontotemporal dementia (FTD), which is characterized by changes in personality and decision-making. Mutations in C9ORF72 can cause ALS or FTD, or even a combination of both, called ALS-FTD. Clumps of TDP-43 proteins accumulate in neurons of people with C9ORF72 mutations, providing another link between the two diseases. This association suggests that ALS and FTD may be part of a series of related conditions, although it is not clear how mutations in the same gene can lead to such different symptoms.
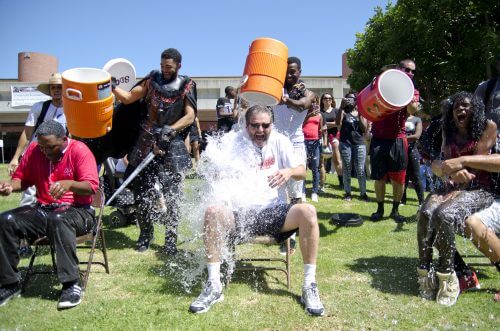
Researchers are looking at three cellular mechanisms that may explain how the mutations in this mysterious gene cause ALS. The repetitive DNA segment can interfere with the way the genetic code is transcribed into messenger RNA and then translated into the C9ORF72 protein, thus reducing the amount of protein produced. This reduction may impair the effects of the protein, although its exact action is still unknown. Alternatively, there could be toxic excess activity: it is possible that the repetitive sequence causes the RNA itself to form clumps that accumulate in the nuclei of nerve cells, acting like a trap for RNA-binding proteins and preventing them from performing their normal tasks. Or there may be toxic excess activity due to some strange disruption of molecular biology where the repeat sequences are translated into small defective proteins that themselves tend to accumulate in the neurons of people with C9ORF7 mutations.
So far, the evidence indicates that mutations in C9ORF7 cause ALS through toxic excess activity, although it is not clear what the relative contribution of RNA blocks is compared to protein blocks. In the end it does not matter, because therapeutic strategies currently in development can silence the production of both the mutant RNA and the mutant protein.
The returns police come to the rescue?
gene silencing using synthetic molecules called antisense oligonucleotides (ASO), is one of the most exciting achievements in the field of degenerative diseases of the nervous system. The ASO molecule is designed so that it locates and binds to a messenger RNA molecule produced by a given gene, and this binding causes the enzyme to go into action and attack the RNA-ASO conjugate. ASO molecules can cause selective destruction of any RNA that is a product of a mutant gene. In the case of C9ORF7, studies done in rodents indicate that antisense molecules engineered to destroy RNA clumps in motor neurons can also destroy clumps of damaged repeat proteins and prevent the formation of new protein clumps.
Antisense drugs that target the C9ORF7 gene are expected to enter human clinical trials this year (2017). Meanwhile, researchers have also designed an antisense drug against the familial form of ALS caused by SOD1, and initial clinical trial results suggest it is safe to use when injected into the cerebrospinal fluid, an injection site chosen to allow the drug to travel through the cerebrospinal fluid that flows around the brain and find its way to the motor neurons.
The success of the ASO drug developed for another degenerative disease of the nervous system, called Spinal muscular atrophy, provides researchers with reason for cautious hope. This genetic disease of motor neurons affects infants and is similar to ALS. Only a few children suffering from the disease live more than three years. In two recent clinical trials with an antisense drug designed to correct the genetic defect that causes the creation of the defective messenger RNA, children with sciatica showed such dramatic improvement in their motor skills that the US Food and Drug Administration (FDA) accelerated the trials and gave official approval for the drug at the end of December 2016.
Solve random ALS
Studies of rare forms of ALS with clear familial inheritance have paved the way for a better understanding of the underlying biology of the disease. The biggest challenge now is to identify mutations that increase disease risk in the genomes of people with sporadic ALS. Around the world, DNA samples of people with ALS are being collected in order to scan their genomes in search of data.
To speed up the search, geneticists have developed a microchip that allows them to conduct genome-wide association studies (GWASs) to easily compare genomes of people with ALS with genomes of healthy people. The chip focuses on regions of the genome known to contain genetic variants called single nucleotide polymorphisms (SNPs), that is, regions where a single DNA signal, or nucleotide, can vary from person to person. GWAS studies are correlational studies, so they cannot find out whether something causes ALS, but they can identify suspicious changes that deserve closer examination. Several international GWAS studies conducted on more than 10,000 people with ALS and more than 20,000 healthy people have revealed some genomic differences that are currently being studied. New technologies have also simplified the process of collecting genetic data, so that a person's complete genome can be sequenced in one day and for less than $1,000. The process is even faster and cheaper if you determine only the sequenceExum, meaning the part of the genome that codes for proteins.
Once researchers have compiled a comprehensive catalog of genetic variants associated with susceptibility to ALS, they will try to decipher the complex ways in which ALS-related mutations increase the risk of the disease. In this rule, they will try to examine how different genes react with each other and if some mutant genes may be involved in some types of ALS, as well as how environmental factors may contribute to the onset of the disease in certain people. Some recent research suggests that ALS may even be caused in part by the reawakening of a dormant retrovirus, a DNA sequence that inserted itself into the genome long ago and usually resides there quietly. It is possible that in some people with ALS, a retrovirus jumps from one nerve cell to another in the brain, causing damage and triggering the disease.
Promising new clues
A growing body of research suggests that ALS is not simply the death of motor neurons. called cells glial cells, which are even more common than neurons in the brain and the central nervous system as a whole, may also play an important role. Glial cells perform a variety of functions: some of them provide physical support to nerve cells; Others regulate the internal environment of the brain, especially the fluid that surrounds nerve cells and their synapses. Recent studies in mice with a mutation in the SOD1 gene yielded a surprise. Stopping the production of the mutant gene in glial cells extended the lives of the mice despite the continued presence of toxic SOD1 proteins in the animals' motor neurons. It seems that the origin of ALS is indeed in the motor neurons, but the communication with the glial cells helps promote the disease. Glial cells may also contribute to ALS by producing a toxic factor, although scientists do not know for sure what this factor is and how it works. Once the cause (or causes) is identified, it will be possible to develop ways to block its production or impair its ability to transmit the harmful signal to motor neurons, in order to slow down or stop ALS.
In the quest to decipher the various causes of ALS, researchers are also trying to identify biomarkers that can help doctors assess the progression of the disease. For example, efforts are being made to identify the proteins containing the excess repeats produced from those expansions of the C9ORF72 gene in available body fluids, such as blood or cerebrospinal fluid. In March 2017 Reporting One of us (Petrocelli) who identified these proteins in the cerebrospinal fluid of people with ALS or with ALS-FTD, as well as in carriers of the mutant gene who have no symptoms. Such tests may help in early diagnosis. Additional studies on biomarkers are focused on developing imaging methods that will help identify clumps of TDP-43 protein that accumulate in the brains of people with ALS, before these clumps begin to kill motor neurons. All these markers may serve as effective indicators for examining the success of possible treatments in clinical trials.
The rapid progress in the fields of genetics and genomics, as well as the development of new and improved biological markers, will bring with it an era of precision medicine against ALS. In the near future, patients will be classified according to their type of ALS and receive treatment or preventive care tailored to them.
The power of social media
Much of the progress in ALS research over the past decade can be attributed to the willingness of a large number of people with the disease to volunteer both their time and their DNA to participate in three large-scale genomic studies. People with ALS and their families have also helped increase public awareness and raise funds to support research and services for patients through the power of social media.
"ALS Ice Bucket Challenge” took the internet by storm in 2014. Pete Freitas, a former captain of the Boston College baseball team who had been diagnosed with ALS two years earlier at age 27, helped spark a stir when he uploaded a Facebook video challenging his friends to flip buckets of ice water over their heads to raise money for ALS Association. The campaign soon went viral and legions of celebrities, including Mark Zuckerberg, Bill Gates, Oprah Winfrey, Leonardo DiCaprio and LeBron James rose to the challenge. Over the course of eight weeks, Facebook users uploaded more than 17 million videos of themselves getting drenched in water for the cause. All participants eventually raised more than 115 million dollars, of which 67% was allocated to research, 20% to services for patients and the community, and 9% to professional and public education.
ASL is a cruel disease that does not let up. Before Gehrig's emotional retirement speech at Yankee Stadium, in which he called himself "the luckiest man on earth," and before the news of his illness became public, most people who had the disease suffered in silence. But now, public awareness continues to grow, thanks in part to people like Freitas. The social media campaign helped revitalize the American ALS Association, which has since tripled its annual research budget. Scientists are optimistic about ALS research, they believe that the rapid growth in understanding the biology of the disease will continue, and that expanding the search for faulty genes will lead to improved treatment that will stop the deadly disease.
- State of Play in Amyotrophic Lateral Sclerosis Genetics. Alan E. Renton, Adriano Chiò and Bryan J. Traynor in Nature Neuroscience, Vol. 17, no. 1, pages 17–23; January 2014
- Decoding ALS: From Genes to Mechanism. J. Paul Taylor, Robert H. Brown, Jr., and Don W. Cleveland in Nature, Vol. 539, pages 197–206; November 10, 2016
- Home page of article author Leonard Petrucelli
- Home page of the author of the article Aaron Gitler

{kind=link}