The gene that causes the severe degenerative disease of the nervous system may have been essential in our evolution

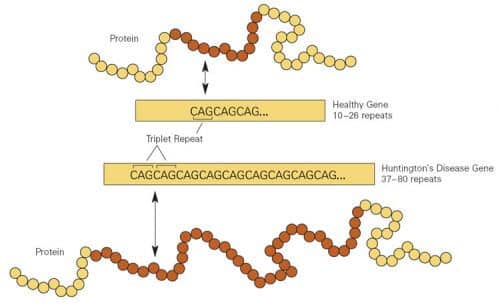
- Huntington's disease, a serious genetic disease that affects the brain, is caused by a mutation in which a trio of DNA letters in the genetic code of the patients repeat themselves too many times.
- Studies have now reconstructed the evolution of the damaged gene. The gene itself first appeared more than a billion years ago and still exists in most species.
- The disease may be an unfortunate byproduct of an evolutionary process. Increasing the number of code letters in a gene probably helped in the development of the nervous system.
- The number of repetitions of the code letters may increase over the generations. A person with too many repeats will develop the symptoms of Huntington's disease.
Starting 15 years ago, British insurance companies agreed not to use the genetic information of policyholders to determine eligibility for life insurance. The dismissal of the claims has one exception. Insurance underwriters can take into account, in certain policies, the fact that the insured carries the gene for a disease that used to be called hereditary chronic chorea and now simply Huntington's disease.
When the insurers are aware of a positive result of the genetic test, they know that since there is still no medical treatment for Huntington's disease, it will most likely be the cause of death of the insured. This knowledge is more certain than other risk factors that insurance companies take into account, such as smoking, consuming alcoholic beverages or riding a motorcycle. A person with a defective gene may experience mood swings and memory disturbances at a young age, usually between the ages of 30 and 50, although these changes can occur later. The symptoms will then worsen, and include involuntary movements, convulsions and an unsteady gait, often described as a kind of fragmented "dancing". Little by little, the body will lose all its abilities and slow down until there is a complete lack of movement, and then the patient will finally succumb to the disease.
Already many years ago, researchers realized that changes in the so-called gene The Huntington They cause disease. We all carry the huntingtin gene because it is important for the development of the nervous system before birth. But there are slight changes in the gene from person to person, and these changes explain why some people will develop Huntington's while the vast majority of others will remain healthy.
One segment of the gene contains a triplet Nucleotides, or "DNA code letters", in this case CAG (Cysteine-Adenine-Guanine) that repeats itself over and over a large number of times. In healthy people, the number of CAG triples ranges from 8 to 35. If the number is higher, the person will eventually get the disease named after George Huntington (1916-1850), the doctor who first described it. One defective copy of the gene (out of the two huntingtin copies that each of us inherits from both parents) is enough to cause the disease. And every child of a sick parent has a high chance of 50% to carry the defective gene. As a result of this inheritance pattern, one out of 10,000 people in Europe and the US gets the disease.
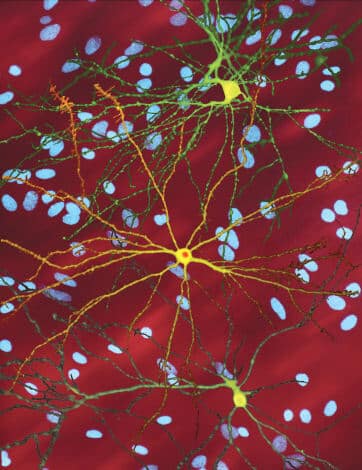
Researchers also knew that the symptoms of Huntington's disease result from the death of nerve cells in two areas of the brain, corpus striatum and the cerebral cortex, which control body movements and high cognitive abilities. Because of this, a significant part of the research on the disease tries to understand how the highly repetitive versions of the gene cause such damage, as well as to develop drugs that will stop the unrestrained progression of the symptoms.
Our laboratory, like many laboratories in different countries, devotes a lot of energy to these goals. A few years ago, during this research, some of us also became interested in the broader question of why harmful versions of genes survive generation after generation and are not weeded out by natural selection. We wondered if this was a kind of biological "walking on the edge" game. Is there an advantage in human survival or reproduction when there are a large, though not too large, number of repetitions? People suffering from the disease also ask this question. They understand that the answer probably won't cure anyone, but they still want to know.
Recently, studies dealing with this question have led to intriguing insights into the role of the gene in the development of the nervous system in humans and other animals. It turns out that an increasing number of CAG repeats contributes, apparently, to the promotion of the function of nerve cells, as long as the increase in the number of repeats does not exceed the threshold of the disease. In this sense, Huntington's disease is not exactly a genetic disease, but may be an unfortunate byproduct of brain design evolution gone awry. A genetic change that may make us "smarter" leads, apparently, to tragic results if it is too extreme. Herein lies the paradox of Huntington's disease.
Genesis
The detective work that led to the understanding of the role played by the gene in the evolution of the nervous system required the researchers to go back in time more than a billion years, to the ancestors of humans and the multicellular amoeba Dictyostelium discoidum. These ancient forms of life lived between the ages ofPaleoproterozoic for the turnMesoproterozoic, and they were the first carriers of the gene, although it was slightly different from the human version.
Descendants of amoebas d. Discoidum still live in soil and leaf rot on the forest floor, and they feed on bacteria. They allowed in 2009 toMiguel Andrade-Navarro, then at the Max Delbrück Center for Molecular Medicine in Berlin, and his group, to search complex databases and find the gene in Amoeba. Andrade-Navarro and his colleagues discovered that one of the differences between the Huntington gene (a less formal name for huntingtin) in amoeba and that in humans is that the gene in amoeba does not contain CAG triplets. Even so, the gene appears to play an important role in one vital step in the organism's life by allowing single-celled amoebae to join together to form the multicellular entity called a pseudoplasmodium.
When there is a shortage of food or when environmental conditions are harsh, this group of amoeba protects itself better than a single amoeba. In 2011, they reported Michael Meyer וJames Gusella from Massachusetts General Hospital that the gene regulates several essential cellular processes, including Dictyostelium's transition to the multicellular stage. Individual cells lacking the Huntington gene have difficulty moving in space and are unable to join other cells in a normal way. It therefore seems that the gene is essential for cells that need to "associate" with each other in order to survive.
In fact, the gene has many functions. A group at Johns Hopkins University discovered that it is responsible for controlling the amoeba's reproduction and their response to environmental stimuli that drive them towards food. In our laboratory, we discovered that the version of the gene found in Dictyostelium protects mammalian cells from environmental stimuli that promote cell death.
More than 550 million years ago, the tree of life split into two branches:protostome, which includes insects, crustaceans and molluscs, and the branchDeuterostome, which led to vertebrates, fish, birds, amphibians, reptiles, mammals, primates and modern man. The amoeba, in which there are no repeats of the three CAGs, precedes the cleavage. After him, only in the deuterostome branch did CAG triplets accumulate at the same point in the gene where the mutations that cause the disease in humans occur.
In 2008, we found that during evolution, CAG triplets began to accumulate in the Huntington gene in animals belonging to the system the skin prickles which belongs to the base of the deuterostome branch, to which, for example, the purple sea urchin belongs, Purpuratus Strongylocentrotus. In collaboration with a group of scientists from our university in Milan, who specialize in computing methods in biology, we decoded the DNA sequence of the gene version found in this sea urchin, and identified two CAG triplets in the first part of the gene.
The DNA sequence in this sea urchin is still different from that of humans. Although sea urchins have a primitive nervous system, the gene is mainly found in non-neural tissues. Its absence suggests that in early stages of evolution, the gene and the two CAG triplets did not play an important role in the nervous system. Research on triplets in the protostome branch is still in its infancy, but it is clear that they are rare (eg, bees have a single CAG). In most cases, these animal systems do not carry CAG in their Huntington gene at all.
In the late 2000s, we analyzed the DNA sequences of the Huntington gene in other deuterosomes in our laboratory, and the most surprising finding was inan awl belonging tothe head strings (which we deciphered with the group of Mario Pastrino from the University of Genoa in Italy). The biology of the eel, a small fish-like creature, marks an important development in the evolution of the nervous system: the acquisition of a polarized nerve structure that stretches from end to end in the animal. The anterior end of the neural cord in Ezmelon has differentiated slightly to form a sort of sac, or vesicle, which may be like a primitive brain.
The DNA sequence showed that, as in sea urchins, here too there are two adjacent CAG triplets. But in this case, the sequence of genetic letters around the pair of triplets was similar to that of vertebrates, including humans, and the protein encoded by the gene is found mainly in nervous tissues. Therefore, we hypothesize that this difference helped create the primitive brain, with its head-tail structure.
When researchers examined the genomes of vertebrates, they found that the CAG triplets begin to lengthen considerably in organisms with more sophisticated nervous systems, and reach their maximum length in humans. This can be inferred when looking at species that are increasingly distant from humans, such as cattle (15 CAG repeats), pigs (18), dogs (10), mice (7) and opossums (6). Individuals of the same species in many organisms, including primates, have CAG segments that vary in length from individual to individual.
Vertebrates mark a new chapter in the evolution of the nervous system. Their brain develops from a hollow structure called neural canal, which is formed in the fetus and then develops into the brain. In 1997, the research group of Marcy McDonald from Massachusetts General Hospital that the Huntington gene is involved in the formation of the neural canal, and in 20122 our group confirmed this finding and expanded upon it after we showed that the gene contributes to the development of a neural canal-like structure in tissue culture.
Human triplets
Meanwhile, other lines of research have begun to chart another role for CAG repeats: brain enhancement. These discoveries are, in part, the result of our efforts that began in the 70s to find the gene that causes Huntington's. Eventually, in 1993, the geneticistNancy Wechsler and 57 other researchers, all inThe Huntington's Disease Collaborative Study Group, isolated and determined the sequence of the human gene, located on chromosome 4, and thus paved the way for the discovery that in people with Huntington's disease the CAG triplets repeat themselves 36 times or more.
A year later he published David S. Rubinstein, a geneticist now working at the University of Cambridge, a paper in which he proposed that the section of the Huntington gene containing the CAG triplets in healthy people tends to expand as it is passed on to offspring. And in 1994, he discovered Max is broke, Nobel laureate from Cambridge, that the amino acid Glutamine, one of the building blocks of proteins, encoded in DNA in the triplet of genetic letters CAG, which repeats itself in the Huntington gene, facilitates the binding to other proteins. However, after these results there was a long hiatus in research about the non-pathological roles of CAG repeats. At the time, CAG repeats and other repetitive DNA sequences were considered genetic "junk", serving no purpose.
In 2008, John V. Fondon the grandson, now at the University of Texas at Arlington, andDavid King From Southern Illinois University Carbondale, they raised a new issue on the subject after they put forward two hypotheses: one, that the nucleotide triplets are involved in the development and evolution of the nervous system, and the second, that increasing the number of triplets in brain cells may improve cognition and the ability to form sexual and social relationships.
Since then, evidence has accumulated to support these hypotheses. A study conducted by the group of Michael Hayden At the University of British Columbia in Vancouver, it was suggested that one person out of every 17 people carries an "intermediate allele", meaning a healthy Huntington's gene with 27 to 35 repeats, a high but not pathological number. In healthy people with a high number of CAG repeats, a tendency to a greater amount of gray matter (nerve cells) was found inglobus pallidus, an area of the brain that controls planning and movement control and is involved in higher cognitive processes. In studies conducted in brain cell cultures, our lab has shown that a greater number of repeats leads to more sophisticated nervous system-like structures [see below].
Even carriers of the gene who are destined to get sick show higher levels of cognitive function. In 2012, they reported Karsten SaftוChristian Best, both then at the Ruhr University in Bochum, Germany, that people with variants of the gene that cause the disease, who have not yet developed symptoms, score better on other visual and perceptual tests than people with normal versions.
brain assistant
New studies on the Huntington's gene also examine the unique tasks that the gene fulfills in the brain. In our research, which was conducted in culture of brain cells in petri dishes, we discovered that the healthy version of the gene makes the nerve cells more resistant to harsh conditions. Conversely, our researchers discovered that silencing the gene in the brains of mice causes cell death and the appearance of symptoms similar to those of mice carrying the harmful version of the gene. We also showed that the gene increases the production of a protein called brain neurotrophic factor (BDNF), which encourages the creation of neural circuits in the brain and the transmission of neural signals.
But perhaps most important of all, the Huntington gene is at its most active in the early stages of embryonic development. Simply put, without him we would not have been born. The gene works during theGastrulation, the stage in embryonic development that begins with the development of the body's main tissues. Later, the gene is responsible for controlling the creation of new nerve cells and helps connect them.
Despite progress, Huntington's paradox still exists. The acquisition of a CAG sequence that continues to extend itself may be the main evolutionary achievement of the Huntington gene, but its tendency to lengthen also poses a dire risk of fatal disease. Questions regarding the repetitive segments in the gene sequence will occupy scientists for many years to come. We still need to better understand why there is such great variation in the number of CAG triplets in a gene. What changes occur in the brain when the number of triplets approaches the threshold that will cause Huntington's disease? Why does the gene suddenly become harmful when there are 36 repeats? The understanding that the Huntington's gene confers both an advantage and a disadvantage can help remove a little of the stigma of the disease, and see it not as a genetic defect but as a product of a biological process that ultimately shaped us as human beings.

{kind=link}
2 תגובות
Just reading a book about it by J. Robert Sawyer, framshift
The author in general likes to give his heroes this disease in a large part of his books 🙁
Correcting a spelling error: the maximum number of repetitions that will not cause illness is 36-35 and not 355 as stated.