Our genome contains tens of thousands of genes, whose expression is controlled by control sequences. In the laboratory of Eran Segal at the Weizmann Institute of Science, they developed a method that allows the study of thousands of such sequences at the same time to obtain a comprehensive picture of how the genome works.
Our genetic information is encoded in long DNA molecules, called chromosomes, which consist of different combinations of four structural units, marked with letters: A, C, G and T. In total, there are about 3.2 billion letters in the genome of each and every one of us. Within these billions of letters are hidden sequences that contain the code for building proteins. However, cell proteins are not produced in the cell nucleus, where the DNA is spun, but in the cell body, called the cytoplasm. If so, how does the information from the DNA in the nucleus reach the cytoplasm? Each gene is transcribed, as needed, into a certain type of RNA molecule, known as messenger RNA, which also consists of four letters, A, C, G and U. RNA, unlike DNA, is able to leave the nucleus to the cytoplasm, where the information encoded in it is translated into proteins by molecular machines called ribosomes. This is the "central dogma" of molecular biology, and for quite a few years it was also the only dogma. In 2000, most of the sequence of letters of the human genome was decoded, and it became clear that about 98% of our genome does not code for proteins. In a certain lack of attention, a significant part of this DNA was called junk DNA, and the general assumption was that it contained evolutionary remnants of past events, such as the penetration of DNA viruses or RNA viruses into the genome of our ancestors or the duplication of DNA segments. A. Because of errors in genome replication.
However, molecular and computational methods that gradually became more sophisticated, also clarified several other things. For example, in this DNA is hidden a whole world of genes that are not translated into proteins, but only copied into RNA, and that these genes have essential roles in controlling other genes (see page 22, as well as "The Unseen Genome: Diamonds in the Junk" by W. White Gibbs, Scientific American Israel issue February-March 2004). Also, the understanding that the genome is a living and breathing environment that responds to a multitude of environmental signals that affect the cell has become more and more acute. For example, the rate of production of each and every gene in our genome is controlled by DNA sequences called promoters. Genes that code for proteins or RNA that are needed in large quantities in the cell will have strong promoters, while other genes, whose products are needed sparingly, will have much weaker promoters. The ability to get to know the various promoters in depth and to understand what affects them is essential so that we can, one day, accurately influence the expression of various genes in our body or in other organisms.
Mathematical models for transcriptional control
The size of the genome, the complexity of the level of expression of the various genes, the enormous amounts of information that are increasing every day that scientists accumulate, and the encrypted nature of the genetic information, all of these are amenable to mathematical treatment that will bring some order to the mess and allow new biological insights. Professor Eran Segal, from the Department of Computer Science and Applied Mathematics at the Weizmann Institute of Science, and his staff, have developed methods that enable the study of the gene control process in a quantitative mathematical way. Segal completed a bachelor's degree in computer science and mathematics at Tel Aviv University, moved to Stanford University for doctoral studies, and did a post-doctoral internship at Rockefeller University in New York. His training process puts him in the right position to deal with the vast amounts of information required to better understand that teeming universe of DNA, RNA and protein molecules that ultimately determine how each cell in our body will look and function. Segal's laboratory focuses on two main topics, one, gene expression and gene control, and the other, a personalized nutrition project, conducted in collaboration with Dr. Eran Alinev, in which they try to understand the interrelationships between our gut bacteria and our diet*.
The goal of Segal's research in the field of transcriptional control is to develop mathematical models that will, in the end, make it possible to predict with great precision the level of expression of each gene based on control sequences in DNA, such as those promoters found at the beginning of genes. To do so, one must first be able to accumulate as much and ordered data as possible on various control sequences that affect gene expression. Data that will make it possible to ask questions such as: which letter sequences within the promoters are the most important for gene control, how does the physical distance between these sequences affect gene expression, how do different environmental conditions affect the operation of the various control sequences, and so on.
reported genes
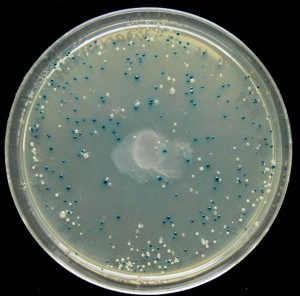
One of the accepted ways to test the effect of different control sequences on gene expression is to connect them to a reporter gene, that is, a gene that encodes a protein whose expression level is easy to follow, such as a fluorescent protein. Thus, if the fluorescent protein is activated by a strong promoter, the cell will glow with strong light when illuminated with light of the correct wavelength. Conversely, if the reporter gene is activated by a weak promoter, the cell expressing that gene will emit weak light. However, in order to collect data that would enable large-scale research on gene control, Segal needed to jump the existing methods a big leap forward. Thus, four years ago, he and his team developed a technique that makes it possible to simultaneously test up to 50,000 different control sequences in 50,000 different cells. In each cell, the reporter gene is attached to a control sequence in which a different mutation has been inserted, and the researchers can follow the effect of each of the 50,000 mutations in the control sequences on the reporter gene and learn how much the different sequences affect the expression level of the protein. Then, and this is where the math comes in, the researchers use the data they received to develop mathematical models capable of describing and predicting the effect of different control sequences on gene expression.
Another direction in which Segal uses this method is the study of the mechanism by which the genetic sequence of viruses, and in particular RNA viruses, whose hereditary information is encoded in RNA molecules, and not in DNA, affects the translation of their proteins in cells. Viruses cannot reproduce on their own, and they need the replication and transcription mechanisms of cells they invade. Thus, new viral proteins are created when the ribosomes of the infected cells translate viral messenger RNA and produce viral proteins based on it. However, in the case of RNA viruses, the translation proceeds slightly differently from the translation of normal messenger RNA. In normal messenger RNA, like human messenger RNA, the ribosome binds to one end of the RNA molecule and starts looking for the starting point of translation. On the other hand, in the case of RNA viruses, the viral RNA molecules have special sequences, called IRES sequences, that direct the ribosome where to start translating. Segal uses his ability to study and analyze thousands of mutations simultaneously to test the effectiveness of thousands of IRES sequences taken from hundreds of different RNA viruses. Here too, the IRES sequences are attached to a reporter gene, and the amount of reporter protein produced indicates the degree of influence the IRES sequence has on gene translation. Among other things, Segal's laboratory will examine how these IRES sequences are affected by short RNA molecules whose role in cells is to inhibit translation of messenger RNA. A good and in-depth understanding of IRES sequences and the translational control of viral proteins is essential for the development of new antiviral drugs.
Personalized medicine for cancer
The beauty of Segal's method is that it can also be applied in completely different directions. For example, another project that the lab is working on is trying to develop personalized medicine to treat cancer tumors that have mutations in the protein called p53, which is responsible, on normal days, for helping damaged cells to commit intentional suicide (apoptosis). Mutations in the p53 gene are among the most common mutations in cancerous tumors. In Segal's laboratory, the effect of thousands of such mutations, isolated from tumors, on the extent of the effect of various chemotherapy treatments on the cells is tested. In this way, it will be possible, in the end, to adapt the correct chemotherapy treatment to the patient, in those cases where there are mutations in the p53 gene.
The combination of biology and mathematics has a long history. Gregor Mendel, the father of modern genetics, already used mathematics and probability to decipher the mode of inheritance of genes. The way in which an electric current develops in nerve cells was deciphered by Alan Hodgkin and Andrew Huxley with the help of a mathematical model, and there are many other examples. But only in recent years, largely due to the genome project, has the understanding been established that without in-depth mathematical treatment, biology will have difficulty moving forward, and in Eran Segal's laboratory this insight is certainly being applied.
Note
The article was published with the permission of Scientific American Israel

2 תגובות
Sherlock
Convergent evolution exists not only in bacteria. Wings evolved at least 4 times, and eyes something like 40 times. It is likely that a "good trick" will evolve several times, and that says nothing about the importance of genes.
I'm not from the field, but from a letter I read recently, about the fact that under the same environmental constraints and the same starting conditions bacteria developed in different evolutionary paths but reached more or less the same features, it seems that genes should be treated like grains of sand in the sea:
Under macroscopic constraints (my hand) it is possible to reach a suitable macroscopic target (a sand castle) even though the microscopic arrangement of the grains can be different.
If we liken the body to a complicated string puppet (it is meant mathematically. Not a hand that is physically tied to something) - the function of the genes is to "grab some strings". Even those that are not related to the same role - the main thing is to catch some (that will have some effect on the macroscopic system). Thus they serve as microscopic degrees of freedom (in the full sense of the term) (the macroscopic change of the body will move the threads and accordingly the genes).
You can try to follow the complicated wire system - start the description of the system from the genes. But if you want a more effective way to influence the system, maybe you need to simulate, somehow, macroscopic constraints... (and speed up the rate of change?...)